Multiple system Atrophy (MSA) defines a specific syndrome within the larger less well defined category of multiple system degenerations. The features of the MSA include: Parkinsonism, cerebellar or corticospinal signs, and autonomic dysfunction (e.g. orthostatic hypotension, impotence, and urinary incontinence or retention), usually preceding or within two years after the onset of the motor symptoms. The previous division into Shy Drager Syndrome (SDS), Striato-Nigral Degeneration (SND), and Olivopontocerebellar Atrophy (OPCA) has been dropped. Immunohistochemistry demonstrates that these three disorders share a common pathology.
The latest diagnostic criteria are shown in the table at the bottom of the page.
Wenning et. al. determined that the combination of autonomic insufficiency, speech or bulbar dysfunction, absence of dementia, postural instability with falls, poor response to levodopa, and absence of levodopa-induced confusion gave a diagnostic sensitivity and specificity greater than 90%.
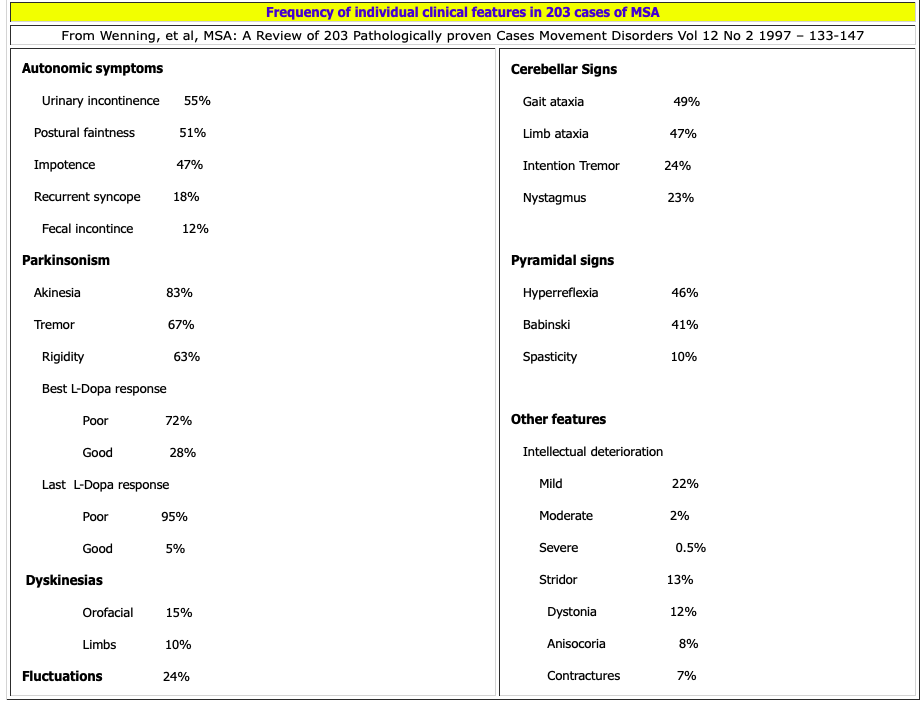
Median age of onset of MSA is about age 55 years (range of 33 to 76). It affects men slightly more than women. Nearly half of patients are disabled or wheelchair bound within 5 years of the onset of motor symptoms. Mean survival is 6 to 7 years. 80% of MSA patients develop predominant Parkinsonism (MSA-P, previously called SND) and 20% develop predominant cerebellar signs (MSA-C, previously called OPCA). There is considerable overlap. Cerebellar features are present in over 40% of patients with SND type, and Parkinsonism is detectable in 50% of OPCA type patients.
30% of MSA-P and 65% of MSA-C patients had a good levodopa response at some stage. Between 13% and 30%, respectively, maintained some response through the course of the illness. 25% to 50% of those treated with levodopa had dyskinesias (particularly orofacial) and dystonia, even if they did not experience improvement in their motor function.
Autonomic symptoms were the initial feature in 41% of patients, but ultimately 97% of patients developed some degree of autonomic dysfunction. The most frequent autonomic symptom in men was impotence and in women urinary incontinence. Orthostatic hypotension occurred in 68%.
A mild restriction of downgaze may develop in about 10% of MSA cases. Anterior horn cell loss may occur but is uncommon. Anal sphincter EMG (90% have an abnormality) is a sensitive and specific diagnostic test for MSA. Even less frequently seen is a mild sensory neuropathy. Classic resting tremor is uncommon (29%). Cerebellar signs occurred in 54% of patients and upper motor neuron signs in 49% of the cases. MSA-P type generally demonstrates more tremor, pyramidal signs, and myoclonus than MSA-C type. Severe dementia is uncommon.
Respiratory stridor (which ultimately occurs in 1/3 of cases) in combination with Parkinsonism is highly suggestive of MSA until proven otherwise; although stridor can also occur in PD, it is exceptionally rare.
Current consensus criteria for the diagnosis of Multiple System Atrophy, adapted from (Gilman et al., 2008).
| Criteria for definite MSA include neuropathological findings during postmortem examination of: |
| a) Widespread and abundant cerebral α-synuclein–positive glial cytoplasmic inclusions, and b) Neurodegenerative changes in striatonigral or olivopontocerebellar region |
| Criteria for probable MSA include a sporadic progressive adult (> 30 years old)–onset disease characterized by: |
| a) Autonomic failure involving urinary incontinence (inability to control the release of urine from the bladder with erectile dysfunction in males) or an orthostatic decrease of blood pressure within 3 min of standing by at least 30 mmHg systolic or 15 mmHg diastolic, and |
| b) Poorly levodopa-responsive parkinsonism (bradykinesia with rigidity, tremor or postural instability), or |
| c) A cerebellar syndrome (gait ataxia with cerebellar dysarthria, limb ataxia or cerebellar oculomotor dysfunction) |
| Criteria for possible MSA include a sporadic progressive adult (> 30 years old)–onset disease characterized by: |
| a) Parkinsonism (bradykinesia with rigidity tremor or postural instability), or |
| b) Cerebellar syndrome (gait ataxia with cerebellar dysarthria limb ataxia or cerebellar oculomotor dysfunction), and |
| c) At least one feature suggesting autonomic dysfunction (otherwise unexplained urinary urgency frequency or incomplete bladder emptying erectile dysfunction in males or significant orthostatic BP decline that does not meet the level required in probable MSA), and |
| d) At least one of the following features: Babinski sign with hyperreflexia – Stridor – Rapidly progressive Parkinsonism – Poor response to levodopa – Postural instability within 3 years of motor onset – Gait ataxia, cerebellar dysarthria, limb ataxia, or cerebellar oculomotor dysfunction – Dysphagia within 5 year of motor onset – Atrophy on MRI of putamen middle cerebellar peduncle, pons or cerebellum – Hypometabolism on FDG-PET in putamen, brainstem or cerebellum – Presynaptic nigrostriatal dopaminergic denervation on SPECT or PET |
Table 2: Features supporting and not supporting a diagnosis of MSA
| Supporting Features | Non-supporting Features |
| Orofacial dystonia | Classic pill-rolling rest tremor |
| Disproportionate anterocollis | Clinically significant neuropathy |
| Camptocormia and/or Pisa syndrome | Hallucinations (not induced by drugs) |
| Contractures of hands and feet | Onset after age 75 |
| Inspiratory sighs | Family history of ataxia or Parkinsonism |
| Severe dysphonia | Dementia (on DMS-IV) |
| Severe dysarthria | White matter lesions suggesting multiple sclerosis |
| New of increased snoring | |
| Cold hands and feet | |
| Pathological laughter or crying | |
| Jerky, myoclonic postural/action tremor |
For more information and support/resources:
- The MSA Coalition: http://www.multiplesystematrophy.org
- NIH Information page: https://www.ninds.nih.gov/Disorders/Patient-Caregiver-Education/Fact-Sheets/Multiple-System-Atrophy